
«ВЕЙНОВСКИЕ ЧТЕНИЯ»
Редкие заболевания порой трудно диагностировать, особенно если они содержат симптомы сразу нескольких более распространённых болезней, которым и отдается приоритет при выборе диагноза.
Ранняя диагностика лизосомальных болезней накоплений чрезвычайно важна, так как своевременное лечение способно предотвратить необратимые последствия для пациентов. Ферментозаместительная терапия позволяет замедлить прогрессирование болезни, улучшить состояние и качество жизни пациента.
Множественные проявления одного заболевания: неврологические осложнения болезни Фабри
При болезни Фабри, носящей наследственный характер, возникает дефицит или снижение активности фермента лизосом α-галактозидазы. Из-за этого не полностью расщепляются гликосфинголипиды. Промежуточные продукты обмена жиров откладываются в различных органах и тканях, вызывая нарушение их функции. Прежде всего, накопление происходит в эндотелиальных и гладкомышечных клетках сосудов, клетках почек, сердечной мышцы, центральной нервной системе, клетках роговицы. В свою очередь это приводит к почечной или сердечной недостаточности. По словам врача неврологического отделения лаборатории нейрохимии им. д-ра Нестора Чамолеса, члена Аргентинского общества неврологов Хуана Мануэля ПОЛИТЕИ, частота заболевания порой оценивается неверно, поскольку за основу берется классический фенотип заболевания. Однако есть и вариант с поздним дебютом, который происходит в одном случае из 2000 рождений. Поздний вариант имеет либо сердечно-сосудистые симптомы, либо симптомы, связанны с болезнью почек. [1]
Для классического варианта болезни Фабри характерны поражения ЖКТ, ангиокератомы, гипогидроз, поражения роговицы глаз, невропатическая боль. Если говорить о позднем варианте, то в возрасте старше тридцати лет проявляются заболевания, которые могут привести к летальному исходу: протеинурия, терминальное заболевание почек, гипертрофия левого желудочка, аритмия.
Поздний дебют болезни Фабри происходит в одном из 2000 рождений
К периферическим неврологическим проявлениям болезни Фабри можно отнести невропатические боли, дисфункцию ЖКТ, потерю слуха, нарушения вегетативной нервной системы и гипогидроз. К центральным неврологическим проявлениям – инсульты, нейропсихиатрические симптомы и дисфункцию вегетативной нервной системы. [2][3]
Сегодня корректно говорить, что нейропатия Фабри – это дизестезия, а не парестезия, так как связана с вовлечением толстых нервных волокон. Характерные пароксизмы боли встречаются часто, но триггерами являются изменения температуры тела.
Основным механизмом патофизиологии служит вовлечение ганглия тарзального корешка: нейроны внутри ганглия заполнены субстратом, сфинголипидами (рис. 1).
Боль в суставах заставляет пациентов обращаться к ревматологам и получать артрит в качестве диагноза. Но в основе лежит невропатическая боль с вовлечением суставов.
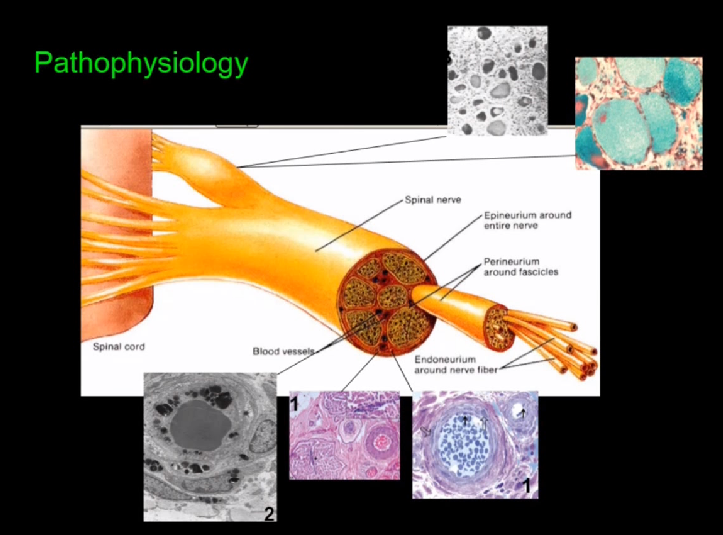
Рис. 1. Механизм патофизиологии
Болезнь Фабри связана с поражением нервов мелких волокон и ганглиев, и поэтому лечить надо не только субстрат, но в первую очередь боль, которая запускается присутствием субстрата. Все начинается с манифестации нарушения вегетативной нервной системы, невропатии малых нервных волокон, нарушений ЖКТ. Надо помнить, что при болезни Фабри бессимптомные инсульты встречаются гораздо чаще, чем манифестные. Важно лечить снижение слуха, предотвращая накопление субстратов в кортиевом органе и в черепно-мозговом нерве. Необходимо стараться избежать развития аритмии, потому что она сопряжена с развитием сердечно-сосудистых и цереброваскулярных кризисов. При своевременной терапии можно предупредить снижение нейрокогнитивных функций у пациентов.
Болезнь Помпе – миопатия о которой нужно знать. Международный опыт диагностики и терапии
Во втором своем докладе врач-невролог Хуан Мануэль Политей раскрыл причины возникновения и методы лечения другой лизосомальной болезни накопления - болезни Помпе. В отличии от болезни Фабри она не связана с Х-сцепленным типом наследования. Причиной заболевания является отсутствие или сниженная активность фермента под названием кислая альфа‐глюкозидаза. Этот фермент необходим для расщепления гликогена до глюкозы в лизосомах. Недостаточная активность этого фермента приводит к внутриклеточному накоплению гликогена. В итоге клетки повреждаются, появляется мышечная дистрофия, мышечная слабость в проксимальных отделах конечностей.
В младенческом возрасте болезнь быстро прогрессирует. Мышечный тонус снижается, дети медленно приобретают или утрачивают моторные навыки. Сердце печень и язык увеличиваются в размерах, развивается парез диафрагмы. В 90% случаев младенцы не доживают до своего первого дня рождения.
Болезнь Помпе с поздним началом отличается более мягкими проявлениями, почти никогда не поражая сердце. Начавшись с незначительной мышечной слабости, она остается незамеченной в среднем в течение 12,6 лет. Это слишком длительный промежуток времени, за который развиваются необратимые изменения в мышечной ткани. Еще до появления мышечной слабости жировая ткань начинает замещать мышечную, что является одним из признаков дистрофии.
Среднее время от первых симптомов до установки диагноза при болезни Помпе составляет 12,6 лет
Каким образом болезнь Помпе диагностируется у людей в позднем возрасте? Из клинических случаев можно рассказать о 52-летней женщине, которую последние 15 лет одолевала мышечная утомляемость и миалгия. Она обращалась к разным специалистам, и все ей ставили диагноз – фибромиалгия. Применяемые анальгетики улучшения не дали. В последний год пациентка стала ощущать слабость в теле, и по этой причине ревматолог решил проконсультироваться с невропатологом. При анализе походки пациентки стало очевидно, что в силу поражения абдоминальных и проксимальных мышц конечностей развился гиперлордоз. Уровень креатинфосфокиназы оказался вдвое выше нормы. Уровень форсированной жизненной ёмкости лёгких снижался более чем на 10% при переходе из положение сидя в положение лежа на спине, что говорило о парезе диафрагмы. По этой причине и был проведен анализ уровня кислой альфа-глюкозидазы методом сухого пятна крови, который оказался сниженным. Далее диагноз подтвердился генетическим анализом – необходимым тестом, который позволяет выявить точные изменения в гене, отвечающего за развитие заболевания [4]
У пациента при поднятии головы в положении лежа пупок смещается в сторону головы. Этот симптом Бивора говорит о мышечной дистрофии. Подпупочные волокна прямой мышцы живота подвержены влиянию болезни Помпе уже на ранних стадиях заболевания (рис. 2). [5]
Оценить слабость языка можно посредством его пальпации. Слабость языка –один из ранних симптомов болезни Помпе. Птоз также является одним из признаков рассматриваемого заболевания. И при сочетании таких симптомов как симптом Бивора, слабость языка и птоз – следует допускать возможность наличия болезни Помпе.
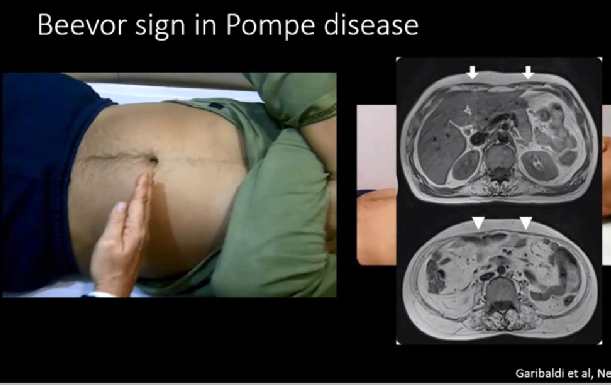
Рис. 2. Симптом Бивора
Оценить мышечную слабость пациента можно посредством динамометра, мышечных и дыхательных тестов.
Болезнь Помпе часто маскируется под симптомами фибромиалгии. Но если они сочетаются с апноэ или одышкой, со снижением уровня форсированной жизненной ёмкости лёгких в положении лежа – необходимо оценить слабость проксимальных мышц.
Путь людей с болезнью Помпе к постановке верного диагноза может занимать годы или даже десятилетия. Это объясняется тем, что в процессе диагностики обычно рассматриваются более распространенные заболевания.
Ранняя диагностика болезни Помпе критически важна, так как без лечения болезнь прогрессирует и приводит к необратимым поражениям органов и систем.
ЛИТЕРАТУРА
- Germain Orphanet Journal of Rare Diseases // 2010, 5:30.
- BMC Neurol // 2011 May. 27:11:61.
- Seroke // 2015 Jan. 46411-302-13.
- European Journal of Neurology // 2017. 0: 1-14.
- Garibaldi et al. Neurology // 2016.
